

La microscopía crioelectrónica rompe una barrera clave que permitirá sondear el funcionamiento de las proteínas con un detalle sin precedentes.
Por Ewen Callaway
Una técnica revolucionaria para la obtención de imágenes de moléculas conocida como microscopía crioelectrónica ha producido sus imágenes más nítidas hasta el momento y, por primera vez, ha discernido átomos individuales en una proteína.
Al lograr la resolución atómica mediante microscopía electrónica criogénica (crio-EM), los investigadores podrán comprender, con un detalle sin precedentes, el funcionamiento de las proteínas que no pueden examinarse fácilmente con otras técnicas de imagen, como la cristalografía de rayos X.
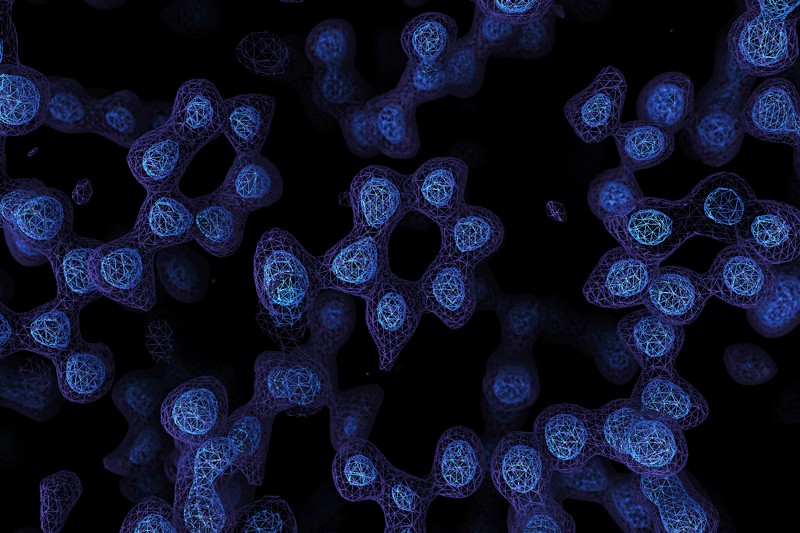
Credit: Paul Emsley/MRC Laboratory of Molecular Biology
El avance, informado por dos laboratorios a fines del mes pasado, consolida la posición de Cryo-EM como la herramienta dominante para mapear las formas 3D de las proteínas, dicen los científicos. En última instancia, estas estructuras ayudarán a los investigadores a comprender cómo funcionan las proteínas en la salud y la enfermedad, y conducirán a mejores medicamentos con menos efectos secundarios.
«Es realmente un hito, eso es seguro. Realmente ya no hay nada que romper. Esta fue la última barrera de resolución», dice Holger Stark, bioquímico y microscopista electrónico del Max Planck Institute for Biophysical Chemistry en Göttingen, Alemania, quien dirigió uno de los estudios. El otro fue dirigido por Sjors Scheres y Radu Aricescu, biólogos estructurales del Medical Research Council Laboratory of Molecular Biology (MRC-LMB) en Cambridge, Reino Unido. Ambos se publicaron en el servidor de preimpresión bioRxiv el 22 de mayo.
«La verdadera ‘resolución atómica’ es un hito real», agrega John Rubinstein, biólogo estructural de la University of Toronto en Canadá. Obtener estructuras de resolución atómica de muchas proteínas seguirá siendo una tarea abrumadora debido a otros desafíos, como la flexibilidad de una proteína. «Estos preprints muestran a dónde se puede llegar si se pueden abordar esas otras limitaciones», agrega.
Rompiendo fronteras
Cryo-EM es una técnica de décadas de antigüedad que determina la forma de muestras ultracongeladas disparándoles electrones y grabando las imágenes resultantes. Los avances en la tecnología para detectar los electrones que rebotan y en el software de análisis de imágenes catalizaron una «revolución de resolución» que comenzó alrededor de 2013. Esto condujo a estructuras de proteínas que eran más nítidas que nunca, y casi tan buenas como las obtenidas con la cristalografía de rayos X, una técnica más antigua que infiere estructuras a partir de patrones de difracción producidos por cristales de proteínas cuando son bombardeados con rayos X.
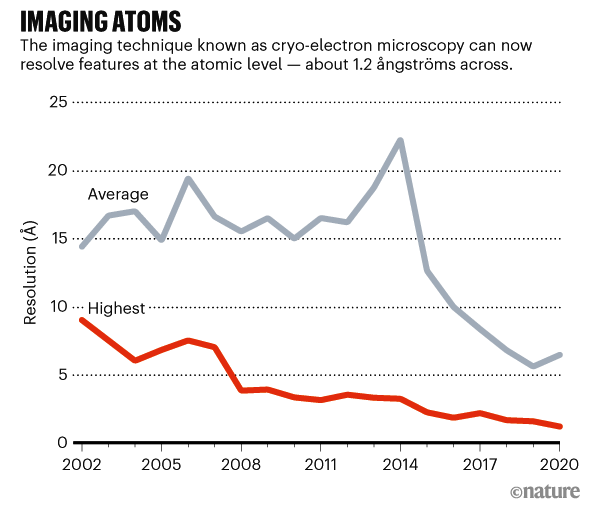
Los avances posteriores de hardware y software llevaron a más mejoras en la resolución de estructuras crio-EM. Pero los científicos han tenido que depender en gran medida de la cristalografía de rayos X para obtener estructuras de resolución atómica. Sin embargo, los investigadores pueden pasar meses o años consiguiendo que una proteína cristalice, y muchas proteínas de importancia médica no formarán cristales utilizables; cryo-EM, por el contrario, solo requiere que la proteína esté en una solución purificada.
Los mapas de resolución atómica son lo suficientemente precisos como para discernir sin ambigüedades la posición de los átomos individuales en una proteína, a una resolución de alrededor de 1,2 ångströms (1,2 × 10-10 m). Estas estructuras son especialmente útiles para comprender cómo funcionan las enzimas y utilizar esos conocimientos para identificar medicamentos que pueden bloquear su actividad.
Para impulsar la crio-EM a una resolución atómica, los dos equipos trabajaron en una proteína de almacenamiento de hierro llamada apoferritina. Debido a su estabilidad similar a una roca, la proteína se ha convertido en un banco de pruebas para crio-EM: una estructura de la proteína con una resolución de 1,54 ångströms fue el récord anterior.
Luego, los equipos utilizaron mejoras tecnológicas para tomar fotografías más nítidas de la apoferritina. El equipo de Stark obtuvo una estructura de 1,25 ångström de la proteína, con la ayuda de un instrumento que asegura que los electrones viajen a velocidades similares antes de golpear una muestra, mejorando la resolución de las imágenes resultantes. Scheres, Aricescu y su grupo utilizaron una tecnología diferente para disparar electrones que viajaban a velocidades similares; también se beneficiaron de una tecnología que reduce el ruido generado después de que algunos electrones salgan de la muestra de proteína, así como una cámara de detección de electrones más sensible. Su estructura de 1,2 ångström era tan completa, dice Scheres, que podían seleccionar átomos de hidrógeno individuales, tanto en la proteína como en las moléculas de agua circundantes.
Stark reconoce que fusionar las tecnologías podría llevar las resoluciones a alrededor de 1 ångström, pero no mucho más. «Por debajo de 1 Å es casi imposible alcanzar la crio-EM», dice. Obtener una estructura de este tipo con la tecnología de vanguardia existente llevaría «varios cientos de años de registro de datos y una cantidad no realista de capacidad de procesamiento y almacenamiento de datos», estima su equipo.
Ver claramente
Scheres y Aricescu también probaron sus mejoras en una forma simplificada de una proteína llamada receptor GABAA. La proteína se encuentra en la membrana de las neuronas y es un objetivo de los anestésicos generales, medicamentos para la ansiedad y muchos otros medicamentos. El año pasado, el equipo de Aricescu utilizó cryo-EM para mapear la proteína a 2,5 ångströms. Pero con el nuevo kit, los investigadores lograron una resolución de 1,7 ångström, con una resolución aún mejor en algunas partes clave de la proteína. «Fue como quitarse una mancha sobre los ojos», dice Aricescu. «Con esta resolución, cada medio ångström abre un universo entero».
La estructura reveló detalles nunca antes vistos en la proteína, incluidas las moléculas de agua en el bolsillo donde se encuentra una sustancia química llamada histamina. «Esa es una mina de oro para el diseño de fármacos basado en estructuras», dice Aricescu, porque muestra cómo un fármaco podría desplazar las moléculas de agua, lo que podría dar como resultado medicamentos con menos efectos secundarios.
Un mapa de resolución atómica de GABAA, que no es tan estable como la apoferritina, sería un desafío, dice Scheres. «No creo que sea imposible, pero sería muy poco práctico», debido a la gran cantidad de datos que sería necesario recopilar. Pero otras mejoras, particularmente en cómo se preparan las muestras de proteínas, podrían allanar el camino para las estructuras de resolución atómica de GABAA y otras proteínas biomédicamente importantes. Las soluciones de proteínas se congelan en pequeñas rejillas hechas de oro, y las alteraciones a estas rejillas podrían mantener las proteínas aún más quietas.
«Todo el mundo está muy emocionado y asombrado por el nivel verdaderamente asombroso de rendimiento demostrado por los grupos MRC-LMB y Max Planck», dice Radostin Danev, especialista en crio-EM de la Universidad de Tokio. Pero está de acuerdo en que la preparación de muestras es el mayor desafío del campo para proteínas más inestables. «El rendimiento de resolución sub-1,5 Å, o incluso sub-2 Å, seguirá siendo accesible durante algún tiempo solo para muestras con buen comportamiento», dice.
Es probable que los avances cimenten la posición de Cryo-EM como la herramienta de referencia para la mayoría de los estudios estructurales, dice Scheres. Las compañías farmacéuticas, que codician las estructuras de resolución atómica, podrían ser incluso más propensas a recurrir a la crio-EM. Pero Stark cree que la cristalografía de rayos X conservará algo de atractivo. Si una proteína se puede cristalizar es relativamente eficiente para generar estructuras unidas a miles de fármacos potenciales en un corto período de tiempo. Pero aún puede llevar horas o días generar suficientes datos para estructuras crio-EM de muy alta resolución.
«Aún existen pros y contras para cada una de las técnicas», dice Stark. «La gente ha publicado muchos artículos y reseñas que dicen que estos últimos avances en crio-EM serán la señal de muerte para los rayos X. Lo dudo».
Referencias
1. Yip, K. M., Fischer, N., Paknia, E., Chari, A. & Stark, H. Preprint at bioRxiv https://doi.org/10.1101/2020.05.21.106740 (2020).
2. Nakane, T. et al. Preprint at bioRxiv https://doi.org/10.1101/2020.05.22.110189 (2020).
3. Kato, T. et al. Microsc. Microanal. 25(S2), 998–999 (2019).
4. Uchański, T. et al. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/812230v1 (2020).
5. Naydenova, K., Peet, M.J. & Russo, C. J. Proc. Natl Acad. Sci. USA 116, 11718–11724 (2019).
Publicado originalmente en Nature
