

Los científicos están desentrañando el ciclo de vida del SARS-CoV-2 y cómo el virus usa trucos para evadir la detección.
El coronavirus luce una lujosa capa de azúcar. «Es sorprendente», pensó Rommie Amaro, mirando su simulación por computadora de una de las proteínas de pico de marca registrada del SARS-CoV-2, que sobresale de la superficie del virus. Estaba envuelto en moléculas de azúcar, conocidas como glicanos.
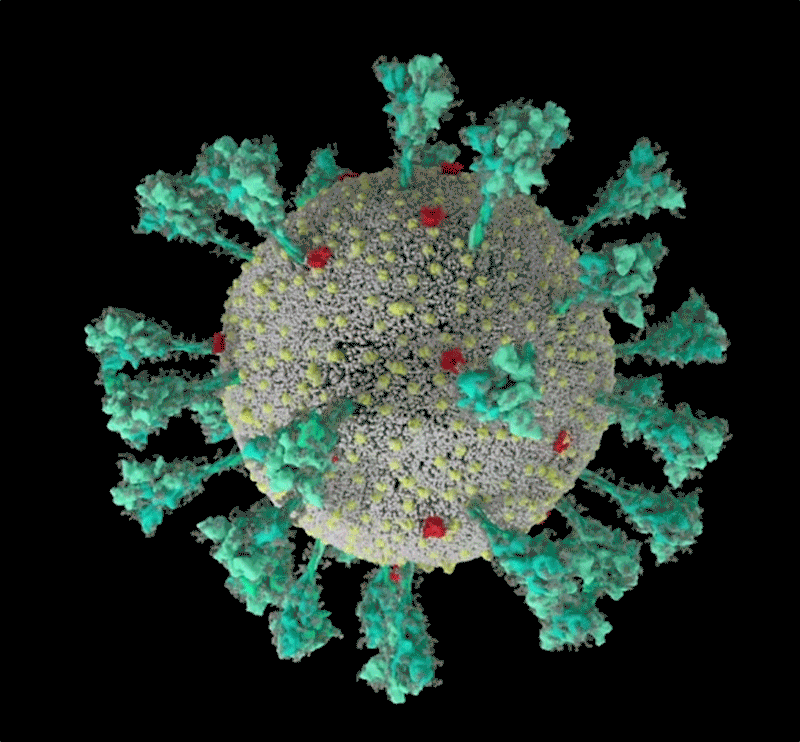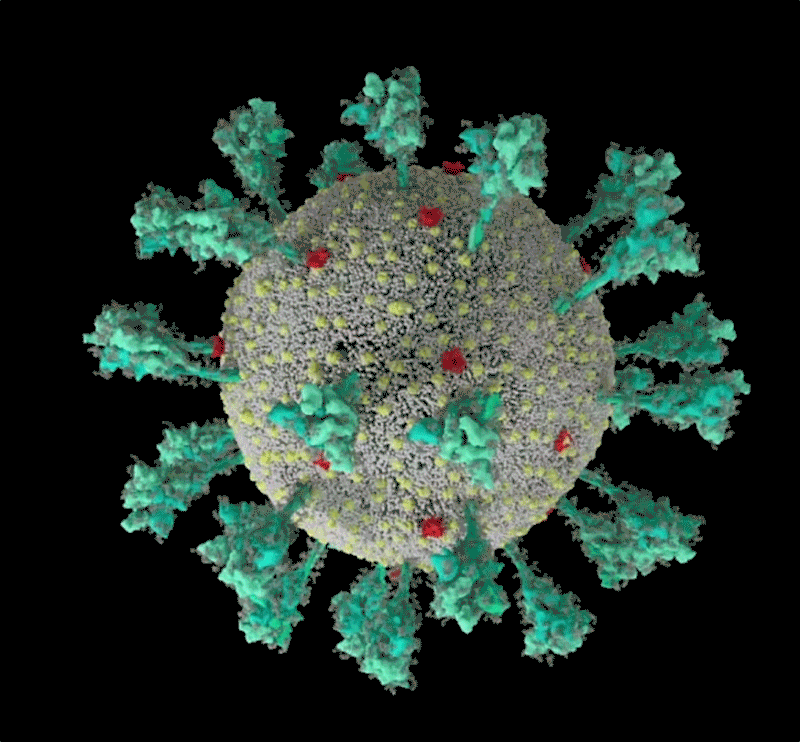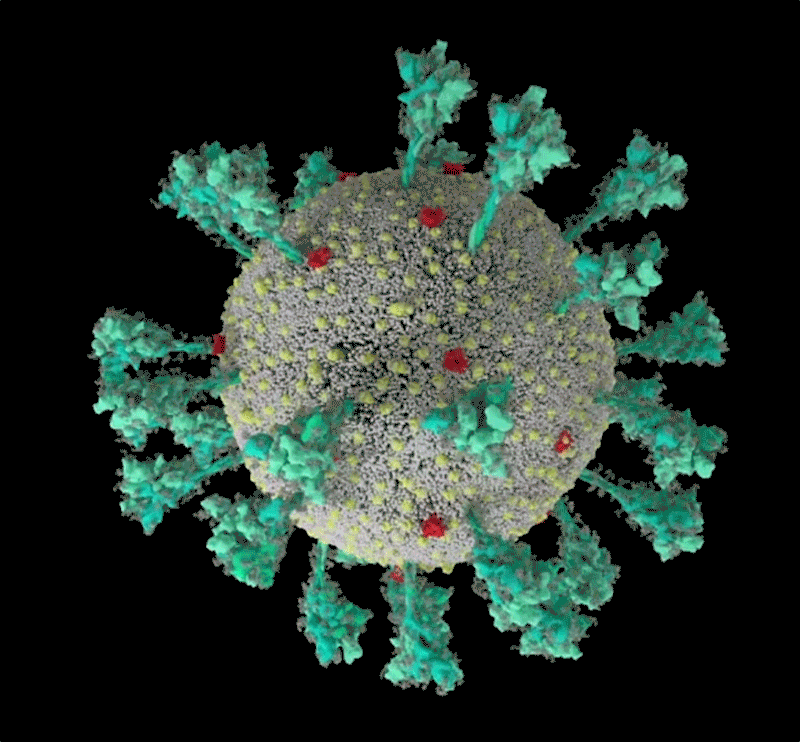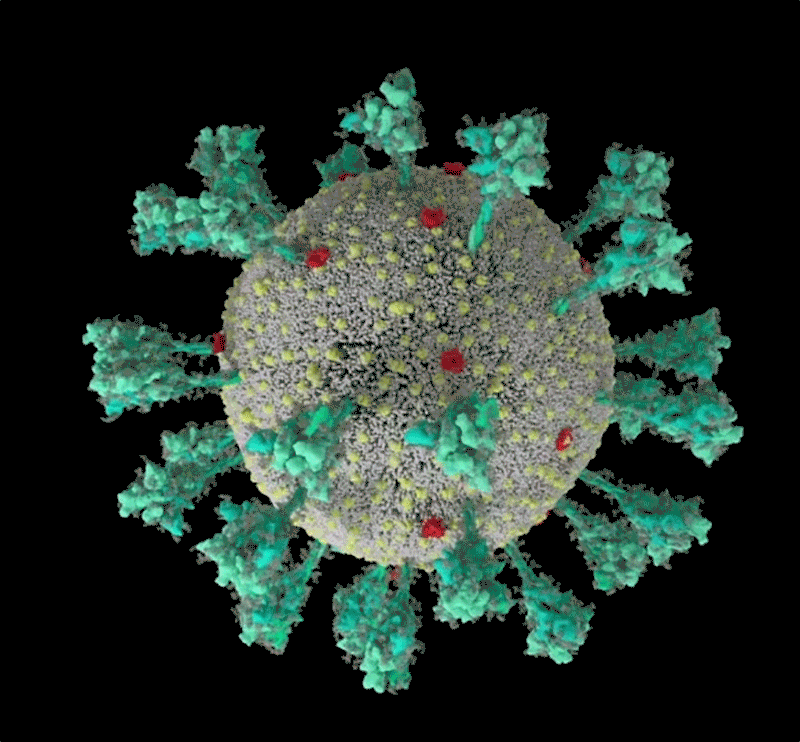
Crédito: Janet Iwasa, Universidad de Utah
«Cuando lo ves con todos los glucanos, es casi irreconocible», dice Amaro, Químico Biofísico Computacional de la Universidad de California en San Diego.
Muchos virus tienen glucanos que cubren sus proteínas externas, camuflándolos del sistema inmunológico humano como un lobo con piel de oveja. Pero el año pasado, el grupo de laboratorio de Amaro y sus colaboradores crearon la visualización más detallada hasta ahora de esta capa, basada en datos estructurales y genéticos y renderizada átomo por átomo por una supercomputadora. El 22 de marzo de 2020, publicó la simulación en Twitter. En una hora, un investigador preguntó en un comentario: ¿qué era el lazo desnudo y sin recubrimiento que sobresalía de la parte superior de la proteína?.
Amaro no tenía idea. Pero diez minutos más tarde, el Biólogo Estructural Jason McLellan de la Universidad de Texas en Austin intervino: el bucle sin recubrimiento era un dominio de unión al receptor (RBD), una de las tres secciones del pico que se unen a los receptores en las células humanas.

Gráfico: Nik Spencer / Nature
En la simulación de Amaro, cuando el RBD se elevó por encima de la nube de glucanos, dos glucanos se abalanzaron para bloquearlo en su lugar, como el soporte de una bicicleta. Cuando Amaro mutó los glucanos en el modelo informático, el RBD colapsó. El equipo de McLellan construyó una forma de probar el mismo experimento en el laboratorio, y en junio de 2020, los colaboradores habían informado que la mutación de los dos glucanos redujo la capacidad de la proteína de punta para unirse a un receptor de células humanas, un papel que nadie había reconocido previamente en los coronavirus, dice McLellan. Es posible que eliminar esos dos azúcares podría reducir la infectividad del virus, dice Amaro, aunque los investigadores aún no tienen una forma de hacerlo.
Desde el comienzo de la pandemia de COVID-19, los científicos han estado desarrollando una comprensión detallada de cómo el SARS-CoV-2 infecta las células. Al separar el proceso de infección, esperan encontrar mejores formas de interrumpirlo mediante tratamientos y vacunas mejorados, y aprender por qué las últimas cepas, como la variante Delta, son más transmisibles.
Lo que ha surgido de 19 meses de trabajo, respaldado por décadas de investigación sobre el coronavirus, es un relato paso a paso de cómo el SARS-CoV-2 invade las células humanas. Los científicos han descubierto adaptaciones clave que ayudan al virus a adherirse a las células humanas con una fuerza sorprendente, y luego esconderse una vez dentro. Más tarde, cuando abandona las células, el SARS-CoV-2 ejecuta un paso de procesamiento crucial para preparar sus partículas para infectar aún más células humanas. Estas son algunas de las herramientas que han permitido que el virus se propague tan rápidamente y se cobra millones de vidas. «Por eso es tan difícil de controlar», dice Wendy Barclay, viróloga del Imperial College de Londres.
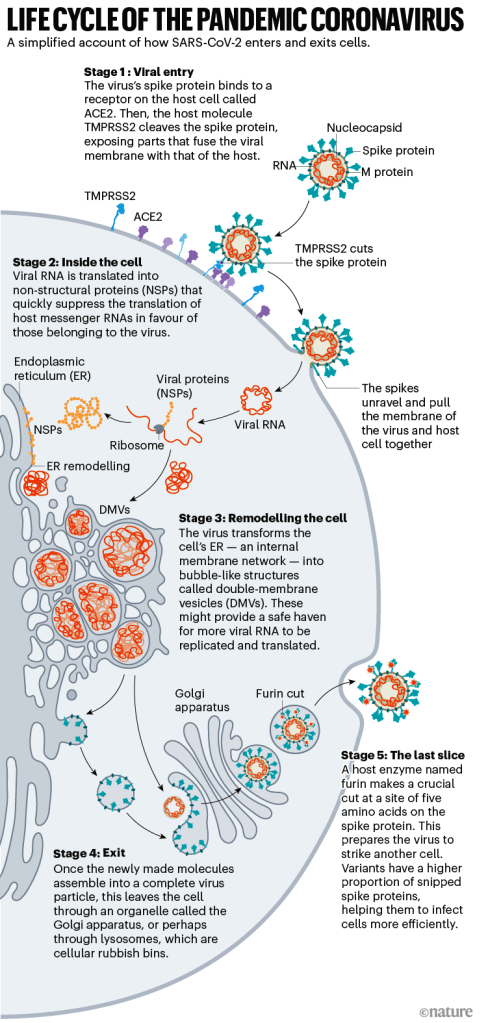
Gráfico: Nik Spencer / Nature
Con púas y listo
Comienza con los picos. Cada virión (partícula de virus) del SARS-CoV-2 tiene una superficie exterior salpicada de 24 a 40 proteínas de punta dispuestas al azar que son la clave para fusionarse con las células humanas. Para otros tipos de virus, como la influenza, las proteínas de fusión externas son relativamente rígidas. Los picos del SARS-CoV-2, sin embargo, son tremendamente flexibles y se articulan en tres puntos, según un trabajo publicado en agosto de 2020 por el bioquímico Martin Beck del Instituto de Biofísica Max Planck en Frankfurt, Alemania, y sus colegas.
Eso permite que los picos se muevan, se balanceen y giren, lo que podría facilitarles la exploración de la superficie celular y que varios picos se unan a una célula humana. No hay datos experimentales similares para otros coronavirus, pero debido a que las secuencias de proteína de punta están altamente conservadas evolutivamente, es correcto asumir que el rasgo es compartido, dice Beck.

Crédito: B. Turoňová et al. / Ciencia
Al comienzo de la pandemia, los investigadores confirmaron que los RBD de las proteínas de pico del SARS-CoV-2 se unen a una proteína conocida llamada receptor ACE2, que adorna el exterior de la mayoría de las células de la garganta y los pulmones humanos. Este receptor también es el punto de acoplamiento del SARS-CoV, el virus que causa el síndrome respiratorio agudo severo (SARS). Pero en comparación con el SARS-CoV, el SARS-CoV-2 se une a ACE2 de 2 a 4 veces más fuertemente, debido a que varios cambios en el RBD estabilizan sus puntos críticos de unión al virus.
Las variantes preocupantes de SARS-CoV-2 tienden a tener mutaciones en la subunidad S1 de la proteína de pico, que alberga los RBD y es responsable de la unión al receptor ACE2. (Una segunda subunidad de pico, S2, provoca la fusión viral con la membrana de la célula huésped).
La variante Alfa, por ejemplo, incluye diez cambios en la secuencia de la proteína de la punta, lo que hace que sea más probable que los RBD permanezcan en la posición «arriba». «Está ayudando al virus al facilitar su entrada en las células», dice Priyamvada Acharya, biólogo estructural del Instituto de Vacunas Humanas de Duke en Durham, Carolina del Norte, que está estudiando las mutaciones de pico.
La variante Delta, que ahora se está extendiendo por todo el mundo, alberga múltiples mutaciones en la subunidad S1, incluidas tres en el RBD que parecen mejorar la capacidad del RBD para unirse a ACE2 y evadir el sistema inmunológico.
Entrada restringida
Una vez que los picos virales se unen a ACE2, otras proteínas en la superficie de la célula huésped inician un proceso que conduce a la fusión de las membranas viral y celular.
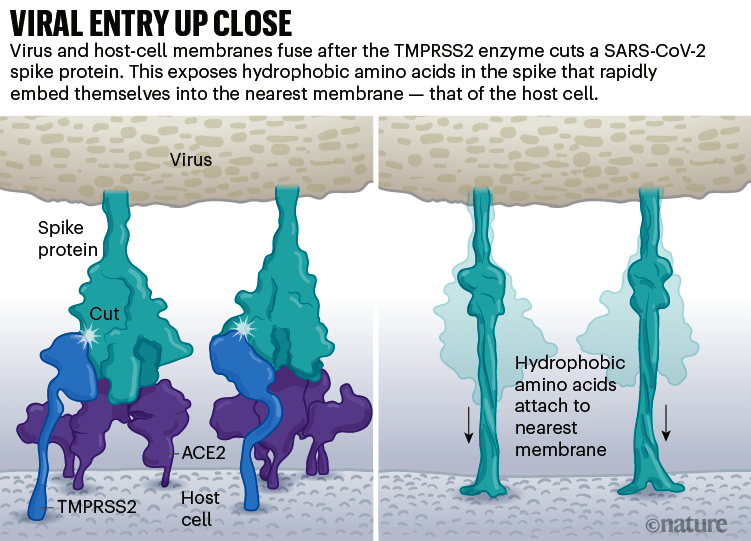
Gráfico: Nik Spencer / Nature
El virus que causa el SARS, SARS-CoV, utiliza cualquiera de las dos enzimas proteasas del huésped para penetrar: TMPRSS2 (pronunciado ‘tempress two’) o catepsina L. TMPRSS2 es la ruta más rápida, pero el SARS-CoV a menudo ingresa a través de un endosoma -una burbuja rodeada de lípidos- que depende de la catepsina L. Sin embargo, cuando los viriones ingresan a las células por esta ruta, las proteínas antivirales pueden atraparlos.
El SARS-CoV-2 se diferencia del SARS-CoV porque utiliza eficientemente TMPRSS2, una enzima que se encuentra en grandes cantidades en el exterior de las células respiratorias. Primero, TMPRSS2 corta un sitio en la subunidad S2 del pico. Ese corte expone una serie de aminoácidos hidrófobos que se entierran rápidamente en la membrana más cercana, la de la célula huésped. A continuación, la espiga extendida se vuelve a doblar sobre sí misma, como una cremallera, obligando a las membranas viral y celular a fusionarse.
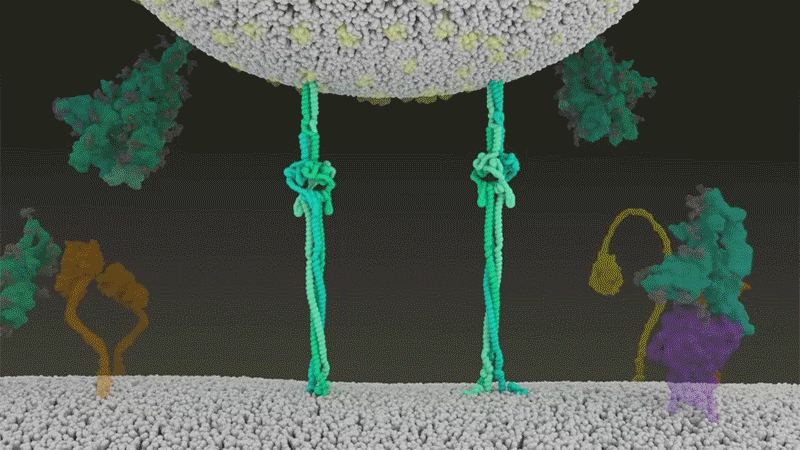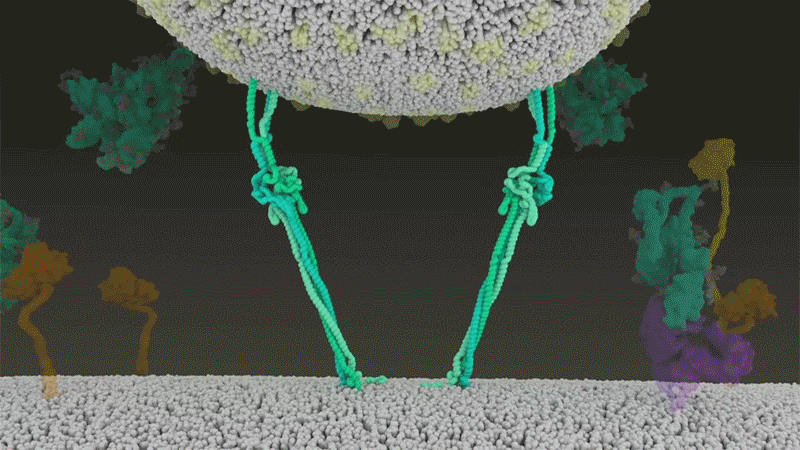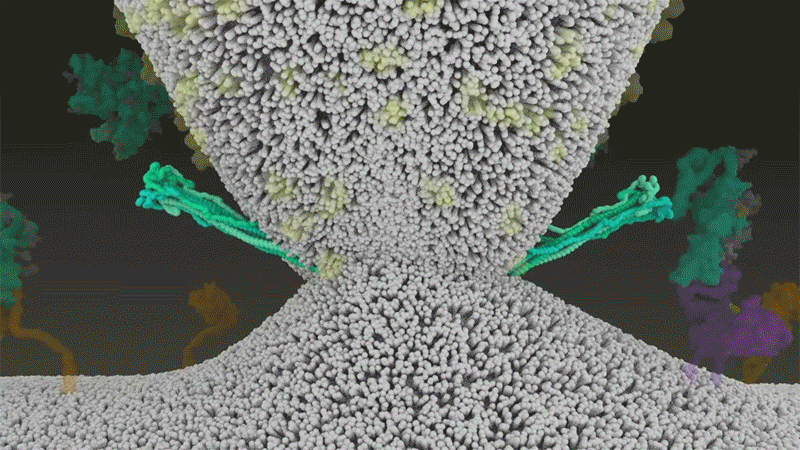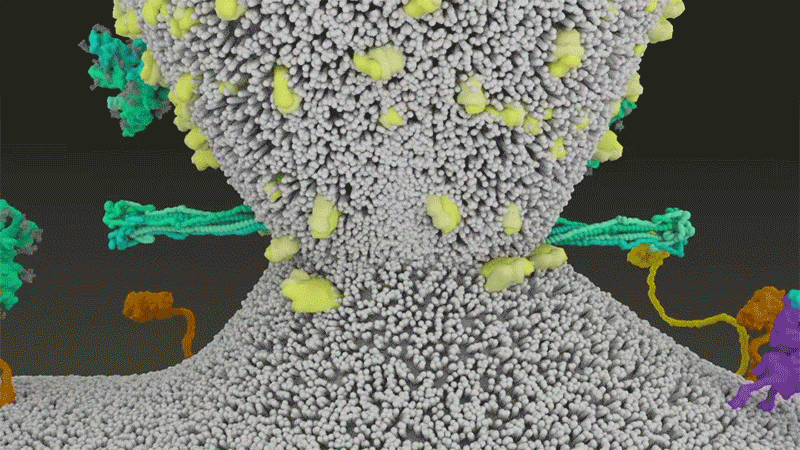
Crédito: Janet Iwasa, Universidad de Utah
Luego, el virus expulsa su genoma directamente a la célula. Al invadir de esta manera cargada por resorte, el SARS-CoV-2 infecta más rápido que el SARS-CoV y evita quedar atrapado en los endosomas, según un trabajo publicado en abril por Barclay y sus colegas en el Imperial College London.
La rápida entrada del virus mediante TMPRSS2 explica por qué el fármaco contra la malaria cloroquina no funcionó en ensayos clínicos como tratamiento para COVID-19, a pesar de los primeros estudios prometedores en el laboratorio. Aquellos resultaron haber usado células que dependen exclusivamente de catepsinas para la entrada endosomal. «Cuando el virus se transmite y se replica en las vías respiratorias humanas, no utiliza endosomas, por lo que la cloroquina, que es un fármaco disruptor endosómico, no es eficaz en la vida real», dice Barclay.
El descubrimiento también apunta a los inhibidores de proteasa como una opción terapéutica prometedora para evitar que un virus use TMPRSS2, catepsina L u otras proteasas para ingresar a las células huésped. Un inhibidor de TMPRSS2, el mesilato de camostat, que está aprobado en Japón para tratar la pancreatitis, bloqueó la entrada del virus en las células pulmonares, pero el fármaco no mejoró los resultados de los pacientes en un ensayo clínico inicial.
«Desde mi perspectiva, deberíamos tener inhibidores de proteasa como antivirales amplios disponibles para combatir nuevos brotes de enfermedades y prevenir futuras pandemias desde el principio», dice Stefan Pöhlmann, director de la Unidad de Biología de Infecciones en el Centro Alemán de Primates en Göttingen, quien ha dirigió la investigación sobre la unión de ACE2 y la vía TMPRSS2.
Competencia mortal
Los siguientes pasos de la infección son más turbios. «Hay muchas más cajas negras una vez que estás dentro de la célula», dice la química Janet Iwasa de la Universidad de Utah en Salt Lake City, quien está desarrollando una animación anotada del ciclo de vida viral. «Hay más incertidumbre e hipótesis contrapuestas».
Después de que el virus dispara su genoma de ARN a la célula, los ribosomas en el citoplasma traducen dos secciones de ARN viral en largas cadenas de aminoácidos, que luego se cortan en 16 proteínas, incluidas muchas involucradas en la síntesis de ARN. Posteriormente, se generan más ARN que codifican un total de 26 proteínas virales conocidas, incluidas las estructurales que se utilizan para producir nuevas partículas de virus, como la espiga, y otras proteínas accesorias. De esta manera, el virus comienza a producir copias de su propio ARN mensajero. Pero necesita la maquinaria de la célula para traducir esos ARNm en proteínas.
Los coronavirus se apoderan de esa maquinaria de muchas formas. La viróloga Noam Stern-Ginossar y su equipo del Instituto de Ciencias Weizmann en Rehovot, Israel, se centraron en tres mecanismos mediante los cuales el SARS-CoV-2 suprime la traducción del ARNm del huésped en favor del suyo propio. Ninguno es exclusivo de este virus, pero la combinación, la velocidad y la magnitud de los efectos parecen únicos, dice Stern-Ginossar.
Primero, el virus elimina la competencia: la proteína viral Nsp1, una de las primeras proteínas traducidas cuando llega el virus, recluta proteínas del huésped para cortar sistemáticamente todos los ARNm celulares que no tienen una etiqueta viral. Cuando el equipo de Stern-Ginossar colocó esa misma etiqueta en el extremo de un ARNm del huésped, el ARNm no se cortó.
En segundo lugar, la infección reduce la traducción total de proteínas en la célula en un 70%. Nsp1 es nuevamente el principal culpable, esta vez bloqueando físicamente el canal de entrada de los ribosomas para que el ARNm no pueda entrar, según el trabajo de dos equipos de investigación. La poca capacidad de traducción que queda está dedicada a los ARN virales, dice Stern-Ginossar.
Finalmente, el virus apaga el sistema de alarma de la célula. Esto sucede de muchas maneras, pero el equipo de Stern-Ginossar identificó un mecanismo claro para el SARS-CoV-2: el virus evita que el ARNm celular salga del núcleo, incluidas las instrucciones para las proteínas destinadas a alertar al sistema inmunológico de la infección. Un segundo equipo confirmó este hallazgo y volvió a señalar a Nsp1: la proteína parece bloquear los canales de salida en el núcleo para que nada pueda escapar.
Debido a que las transcripciones de genes no pueden salir del núcleo, las células infectadas no liberan muchos interferones; estas son proteínas de señalización que alertan al sistema inmunológico de la presencia de un virus. El SARS-Cov-2 es particularmente eficaz para apagar este sistema de alarma: en comparación con otros virus respiratorios, incluidos el SARS-CoV y el virus sincitial respiratorio, la infección del SARS-CoV-2 induce niveles significativamente más bajos de interferones. Y este junio, los investigadores informaron mutaciones en la variante Alpha que parecen permitirle controlar la producción de interferón de manera aún más eficiente.
«Está claro que el SARS-CoV-2 es un virus muy rápido que tiene una capacidad única para evitar que nuestro sistema inmunológico reconozca y combata la infección en las primeras etapas», dice Stern-Ginossar. Para cuando el sistema inmunológico se da cuenta de que hay un virus, hay tanto que las proteínas de respuesta inmunitaria a veces inundan el torrente sanguíneo a un ritmo más rápido de lo normal, lo que puede causar daños. Los médicos vieron al comienzo de la pandemia que algunas personas con COVID-19 que se enferman gravemente se ven perjudicadas por una respuesta inmune hiperactiva al SARS-CoV-2, así como por el virus mismo. Algunos tratamientos probados funcionan amortiguando esta respuesta inmune .
Estación de renovación
Una vez que el virus se ha hecho cargo de la traducción del anfitrión, comienza un cambio de imagen en el hogar, remodelando en gran medida el interior y el exterior de la célula según sus necesidades.
Primero, algunas de las proteínas de pico virales recién creadas viajan a la superficie de la célula y sobresalen de la membrana de la célula huésped. Allí, activan un canal de iones de calcio del huésped, que expulsa una capa de grasa hacia el exterior de la célula, la misma capa que se encuentra en las células que se fusionan naturalmente, como las células musculares. En este punto, la célula infectada se fusiona con las células vecinas que expresan ACE2 y se convierte en células respiratorias individuales masivas llenas de hasta 20 núcleos.
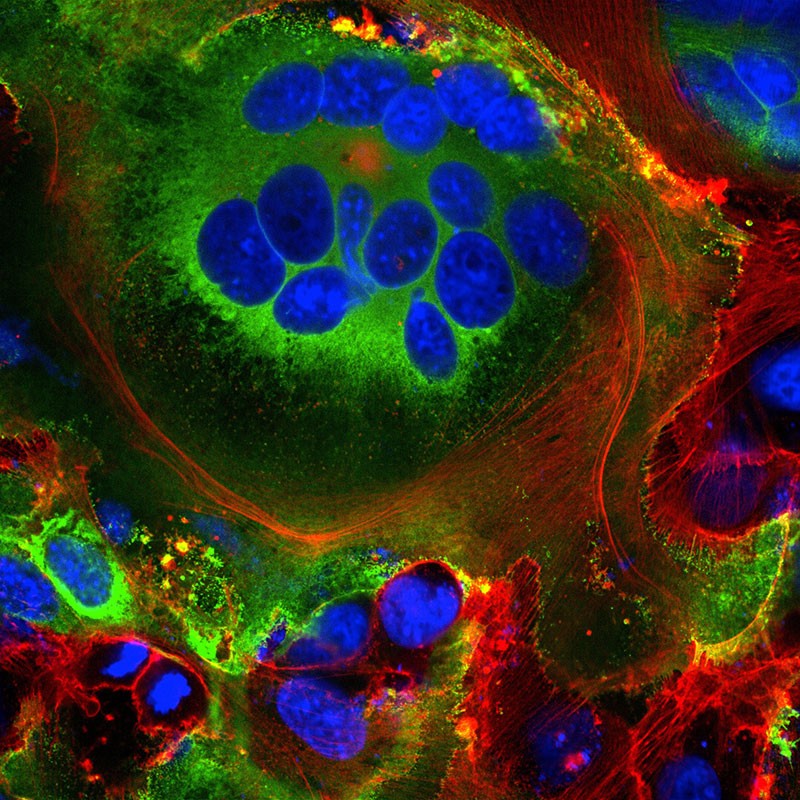
Crédito: Mauro Giacca
Estas estructuras fusionadas, llamadas sincitios, son inducidas por infecciones virales como el VIH y el virus del herpes simple, pero no por el virus del SARS, dice el biólogo molecular Mauro Giacca del King’s College de Londres, quien dirigió el equipo que publicó el hallazgo el 18 de abril. Él plantea la hipótesis de que la formación de sincitios permite que las células infectadas prosperen durante largos períodos de tiempo, produciendo más y más viriones. «Este no es un virus que se da a la fuga», dice. «Persiste». Un segundo equipo, dirigido por el investigador Qiang Sun de la Academia China de Ciencias Médicas en Beijing, descubrió que algunas células infectadas con COVID-19 incluso forman sincitios con linfocitos, una de las células inmunitarias del propio cuerpo. Este es un mecanismo conocido de evasión inmunitaria por parte de las células tumorales, pero no por los virus. Sugiere que las células infectadas evitan la detección inmunológica simplemente agarrándose y fusionándose con exploradores inmunes cercanos.
En el interior de la celda, se están produciendo aún más cambios. Al igual que otros coronavirus, el SARS-CoV-2 transforma el retículo endoplásmico (RE) largo y delgado, una red de membranas planas involucradas en la síntesis y el transporte de proteínas, en esferas de doble membrana, como si el RE estuviera soplando burbujas. Estas vesículas de doble membrana (DMV) podrían proporcionar un lugar seguro para que el ARN viral se replique y traduzca, protegiéndolo de los sensores inmunes innatos en la célula, pero esa hipótesis aún se está investigando.
Las proteínas involucradas en la fabricación de DMV podrían ser buenos objetivos farmacológicos, porque parecen ser necesarias para la replicación viral. Por ejemplo, se necesita una proteína huésped, TMEM41B, para movilizar el colesterol y otros lípidos para expandir las membranas del RE de modo que todas las partes del virus quepan dentro. «Cuando se quita TMEM41B, tiene un gran impacto en la infección», dice Vineet Menachery, investigador de coronavirus en la Rama Médica de la Universidad de Texas en Galveston, que participó en la investigación. La proteína transmembrana del coronavirus Nsp3 también podría ser un objetivo: crea un poro en forma de corona en las paredes de los DMV para lanzar el ARN viral recién creado .
La mayoría de los virus que tienen una envoltura externa, conocida como sobre, forman esta característica al ensamblarse directamente en el borde de la célula, cooptando parte de la propia membrana plasmática de la célula al salir. Pero las proteínas de coronavirus recién creadas toman un camino diferente.
Durante años, la evidencia ha sugerido que los coronavirus se transportan fuera de la célula a través del complejo de Golgi, un orgánulo que funciona como una oficina de correos, empaquetando moléculas en membranas y enviándolas a otras partes de la célula. Allí, el virus forma una envoltura lipídica a partir de la membrana del complejo de Golgi; Los viriones recién formados se transportan dentro de las vesículas de Golgi a la superficie celular, donde son lanzados fuera de la célula, dice la viróloga y bióloga celular Carolyn Machamer de la Universidad Johns Hopkins en Baltimore, Maryland, quien ha estudiado los coronavirus durante 30 años.
Pero en diciembre, la bióloga celular Nihal Altan-Bonnet del Instituto Nacional del Corazón, los Pulmones y la Sangre de EE. UU. En Bethesda, Maryland, y sus colegas informaron que habían detectado coronavirus que salían de la célula a través de lisosomas, contenedores de basura celular llenos de enzimas que descomponen partes de la célula. El bloqueo de la vía secretora basada en Golgi no pareció afectar la cantidad de virus infeccioso que se libera, dice Altan-Bonnet. La evidencia de su equipo sugiere que las proteínas virales forman una envoltura al brotar en el ER y luego se apoderan de los lisosomas para salir de la célula. Actualmente, los investigadores están probando inhibidores que bloquean el proceso de salida lisosomal como posibles candidatos antivirales.
Dejar una célula a través del Golgi o los lisosomas es lento e ineficiente en comparación con la gemación de una membrana plasmática, por lo que los científicos no saben por qué lo hace el SARS-CoV-2. Machamer sospecha que la composición lipídica de una envoltura derivada de Golgi o lisosoma es de alguna manera más beneficiosa para el virus que una de la membrana plasmática. «Si entendiéramos esta parte un poco mejor, habría grandes oportunidades para nuevas terapias antivirales», dice.
Última rebanada
Al salir de la célula, un evento más convierte a este virus en un monstruo infeccioso: un corte rápido en un sitio de cinco aminoácidos prepara al virus para atacar a su próximo objetivo.
Donde otros coronavirus tienen un solo aminoácido de arginina en la unión de las subunidades S1 y S2 del pico, el SARS-CoV-2 tiene una línea de cinco aminoácidos: prolina, arginina, arginina, alanina y arginina. «Debido a que el sitio era inusual, nos enfocamos en él y resultó que sí, el sitio es esencial para la invasión de las células pulmonares», dice Pöhlmann. En mayo de 2020, él y sus colegas informaron que una proteína de la célula huésped llamada furina reconoce y recorta esa cadena de aminoácidos, y el corte es «esencial» para que el virus ingrese a las células pulmonares humanas de manera eficiente.
No es la primera vez que los investigadores han identificado un sitio de división de furina en un virus; los virus de la influenza aviar altamente patógenos también la tienen, dice Barclay. Cuando un colega envió a Barclay una cepa de SARS-CoV-2 en cultivo que había perdido espontáneamente el sitio de división de la furina, su equipo descubrió que los hurones infectados con esta cepa arrojaban partículas virales en cantidades más bajas que los infectados con la cepa pandémica, y no transmitir la infección a los animales cercanos. Al mismo tiempo que el equipo de Barclay informó sus resultados en una preimpresión de septiembre de 2020, un estudio en los Países Bajos también encontró que el coronavirus con un sitio de división de furina intacto ingresa a las células de las vías respiratorias humanas más rápido que aquellos sin él.
Se sospecha que furina corta el sitio en algún momento durante el ensamblaje del virión o justo antes de su liberación. El momento podría explicar por qué el virus sale a través del Golgi o los lisosomas, dice Tom Gallagher, virólogo de la Universidad Loyola de Chicago en Illinois. «El virus, una vez ensamblado, se mueve a un orgánulo donde puede bañarse en presencia de la proteasa furina».
Al cortar el enlace entre las subunidades S1 y S2, el corte de furina afloja las proteínas del pico del virión para que durante la entrada celular respondan a un segundo corte de TMPRSS2, que expone el área hidrofóbica que se entierra rápidamente en una membrana de la célula huésped, dice Gallagher. Si los picos no están precortados por furina, y no siempre lo hacen, pasan por alto TMPRSS2 y entran a través de la vía endosomal más lenta, si es que lo hacen.
Dos variantes de coronavirus, Alfa y Delta, han alterado los sitios de escisión de la furina. En la variante Alfa, el aminoácido prolina inicial se cambia a una histidina (P681H); en la variante Delta, se cambia a arginina (P681R). Ambos cambios hacen que la secuencia sea menos ácida, y cuanto más básica es la cadena de aminoácidos, la furina la reconoce y corta con mayor eficacia, dice Barclay. «Podríamos plantear la hipótesis de que este virus está mejorando aún más su transmisión».
Más cortes de furina significan más proteínas de pico preparadas para ingresar a las células humanas. En el SARS-CoV, menos del 10% de las proteínas de pico están preparadas, dice Menachery, cuyo grupo de laboratorio ha estado cuantificando las proteínas de pico preparadas pero aún no ha publicado este trabajo. En el SARS-CoV-2, ese porcentaje se eleva al 50%. En la variante Alpha, es más del 50%. En la variante Delta altamente transmisible, según el grupo, más del 75% de los picos están preparados para infectar una célula humana.
Desconocidas conocidas
La comunidad científica todavía está rascando la superficie de su comprensión del SARS-CoV-2. Las incógnitas clave incluyen el número de receptores ACE2 necesarios para unirse a cada proteína de pico; cuando exactamente el sitio S2 es escindido por TMPRSS2; y la cantidad de picos necesarios para la fusión de la membrana de la célula y el virus, dice McLellan, y eso es solo para ingresar. En abril de 2020, un equipo de la Universidad de California en San Francisco identificó al menos 332 interacciones entre el SARS-CoV-2 y las proteínas humanas.
No es fácil seguir el ritmo de la rápida mutación del virus. La mayoría de las mutaciones hasta ahora están asociadas con la eficacia con la que se propaga el virus, no con cuánto daña el virus al huésped, según coinciden los expertos. Este mes, un estudio informó que la variante Delta creció más rápidamente y en niveles más altos dentro de los pulmones y la garganta de las personas que las versiones anteriores del virus.
Pero todavía no es seguro cómo las mutaciones de Delta han sobrealimentado la variante de esta manera, dice Stern-Ginossar. «Esto es algo que muchos laboratorios están tratando de resolver».
Fuente: Nature
